
Here you can find a series of very short video tutorials that will guide you through the basic functionalities of the Interactome3D web service.
 |
 |
 |
| Overview | Submit your data and check the results | Browse Interactome3D pre-built datasets |
|---|---|---|
 |
 |
|
| Query with a list of proteins | Download Interactome3D data |
You can also follow our step-by-step tutorials:
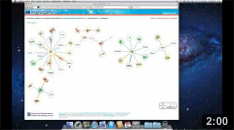
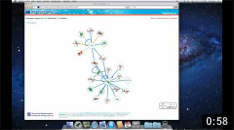
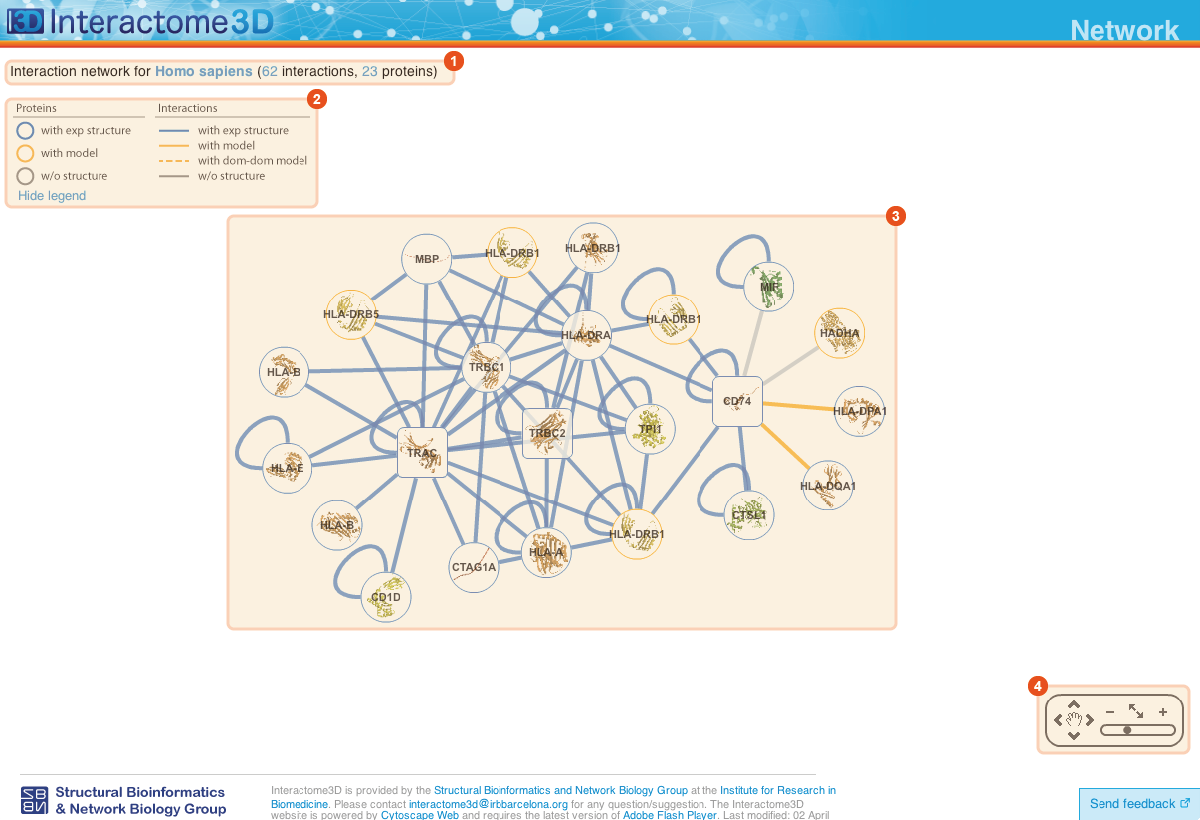
The Network page allows the user to get an overview of the interaction network together with the available structural data for both proteins and interactions and to access the details about these data by clicking on the nodes or on the edges of the network.
![]()
1 |
Title The title at the top of the page summarizes the contents of the displayed network (name of the dataset, organism or GO term selected). In addition it also reports the number of proteins (nodes) and interactions (edges) displayed. |
2 |
Legend Indicates the colors and symbols used in the network view. It can be hidden/shown by clicking on the respective link. |
3 |
Network view Interactive visualization of the network. The nodes represent proteins while the edges represent interactions. A node can be moved with the mouse by drag and drop. Clicking on a node or an edge takes the user to the corresponding protein or interaction page. |
4 |
Tool box The tool box allows to navigate on the network view by moving around and zooming in and out. |
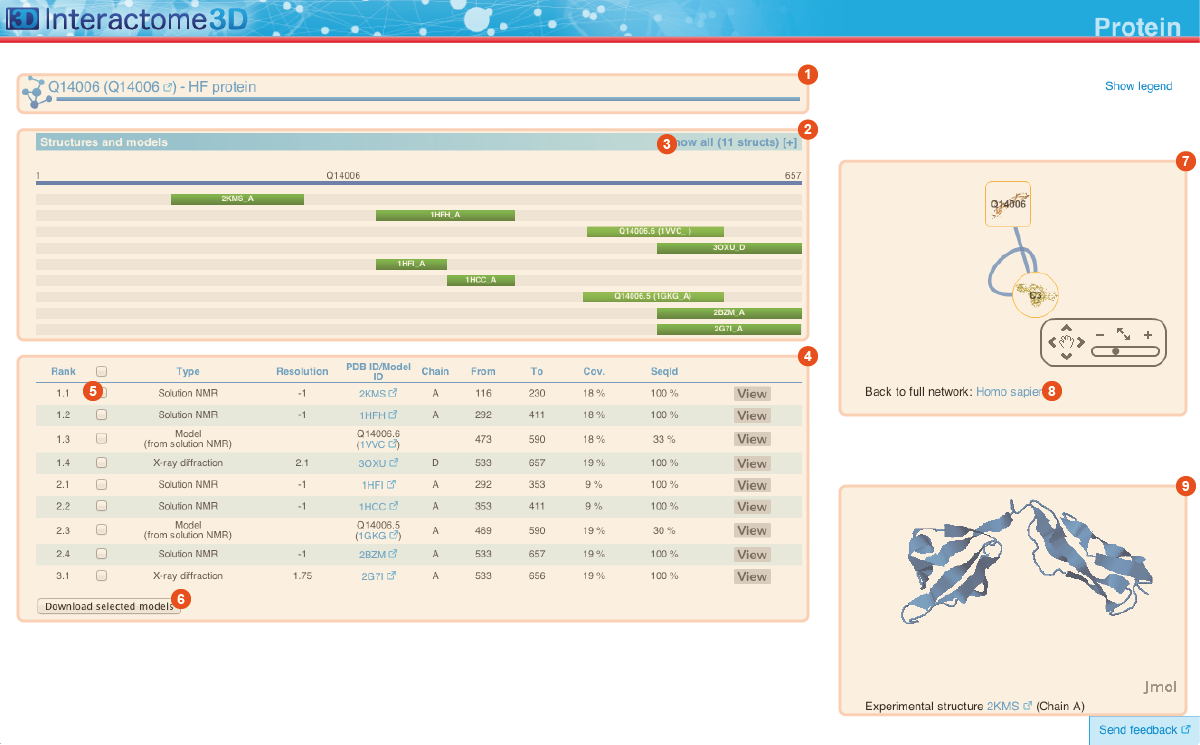
The Protein page displays details about the protein, including all the available structural data and allows the user to preview and download the structures and models.
![]()
1 |
Title The title at the top of the page indicates Gene symbol, the Uniprot AC and the name of the protein. The Uniprot AC links directly to the corresponding entry page in Uniprot. |
2 |
Structures and models visualization pane A graphical pane displays the parts of the protein that are present in the different protein structures/models. Every line represents a different structure/model and the colored bars indicate which portion of the protein sequence is modeled in the structure. The intensity of the color depends on the sequence identity of the template to the protein. For every protein there can be multiple structures/models. Different structures/models can span different portions of the protein. A protein can have all types of structural data: complete experimental structures (covering >80% of the length of the protein with 100% sequence identity), complete homology models (>80% coverage) and partial experimental structures or models (the rest). By hovering over the lines in this pane you get a description of the PDB entry from which the structure or the model were taken. By clicking on a line, the corresponding row will be automatically selected/deselected in the table (4). By default only a set of representative structures and models (top ranked structures and models) are shown. You may use the link (3) to show/hide the other structures/models. |
4 |
Structures and models table This table contains details for the different structures and models for the protein. It reports, for example, the type of structure/model, the PDB ID from which it was taken or from which the template was extracted, the starting and ending modeled residues, etc... By clicking on the View button you can get a preview of the structure in the Jmol view (9) at the bottom right of the page. You can also select a set of models using the checkboxes (5) and download the corresponding PDB files (6). The different structures/models are ranked based on their quality/completeness. Please refer to this description for details on how the ranking is done. |
7 |
Protein neighborhood This pane presents a quick view of the protein neighborhood. It contains all the neighboring interactions that involve the protein. You can browse and click on it just like you do on the network visualization of the Network page. By clicking on the link at the bottom of the network view (8) you can go back to the Network page and visualize the entire network from which you started your browsing (the one you obtained originally by querying Interactome3D with a list of proteins of your interest or by browsing it using GO terms). |
8 |
Jmol view The Jmol applet on the bottom right of the page shows a preview of the structure/model for the protein. In case of multiple structures/models you can change the one that is visualized by clicking on the "View" buttons of the Structures and models table (4). |
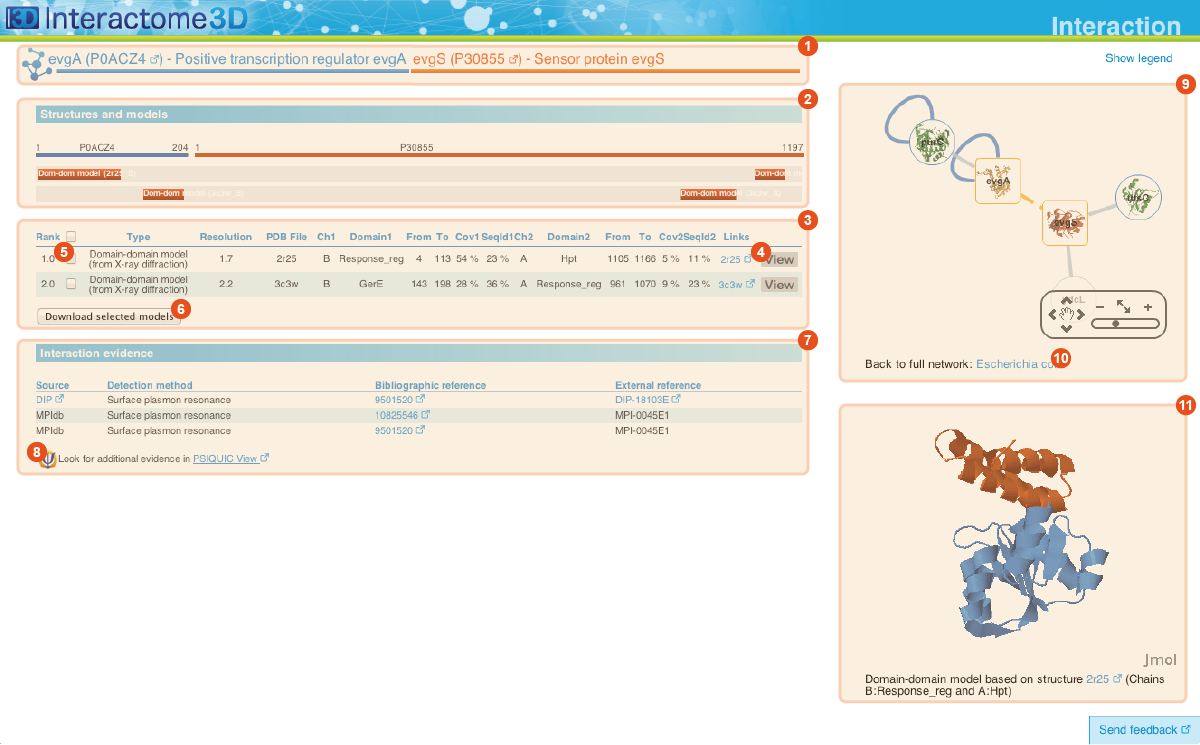
The Interaction page displays details about the interaction, including all the available structural data and allows the user to preview and download the structures and to access external experimental evidences for the interaction.
![]()
1 |
Title The title at the top of the page indicates the two interacting proteins and report their corresponding Gene symbols, the Uniprot ACs and the Protein names. The Uniprot ACs link directly to the corresponding entry page in Uniprot. |
2 |
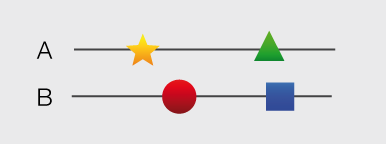
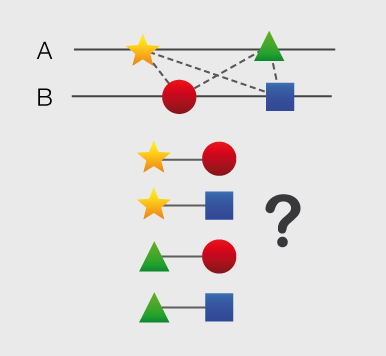
Structures and models visualization pane A graphical pane displays the parts of the two interactors that are present in the different interaction structures/models. Every line represents a different structure/model and the colored bars indicate which portion of the protein sequence is modeled in the structure. The colors indicate the type of the structure/model: green for experimental structures, orange for homology models from global templates and red for models from domain-domain templates. The intensity of the color depends on the sequence identity between the template and the target protein. For every interaction there can be multiple structures/models. Different structures/models for an interaction can span different portions of the two proteins and show different interaction modes. A quick look to this pane will help you in distinguishing between different structural modes of interactions. Any interaction can have only one of the different types of structural data (experimental structures, models from global templates and models from domain-domain structural templates). This means that, if an interaction has experimental structural data, no homology model is produced for it. If there is no experimental structure but there are global templates available than only the models from the gloabal templates are produced and not the models from domain-domain templates. If neither experimental structures nor global templates are available then Interactome3D tries to model the interaction on the basis of the available domain-domain structural templates. The modeling strategy adopted by Interactome3D is better described in the Interactome3D publication (see the references page). Nevertheless, we would like to spend a few words here on the strategy used for the selection of domain-domain structural templates when multiple templates are available. When modeling an interaction using domain-domain structural templates it is possible that the domain assignment to the two proteins allows for multiple possible domain-domain pairs mediating the interaction.
For example, if the star, square, circle and triangle in the previous image all represent different domains then there are at least 4 possible domain-domain pairs that mediate the interaction.
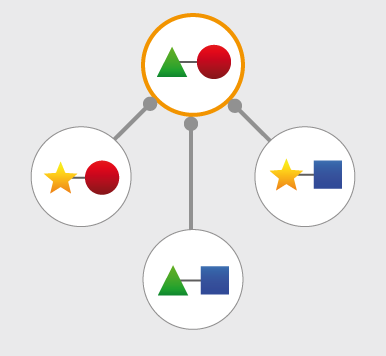
In order to select one structural template for the interaction we need to decide which domain-domain template is the most likely to mediate the interaction. This selection, in Interactome3D, is done using a strategy based on the network of preferred domain-domain interaction pairs observed in the PDB, as described in the work of Itzhaki et al. (Itzhazi, Akiva & Margalit, Bioinformatics 26:2564-2570, 2010). Given all the possible domain-domain pairs we build the network of preference between them and we select the one that dominates over the others. Please refer to the original publication for a description of how the network of preference is built.
Then we select in 3did all the structural templates corresponding to this particular domain-domain pair and we take the template corresponding to the most numerous topology in 3did. There is still the possibility that no domain-domain pair dominates over the others. This happens when the network of preference forms a cycle.
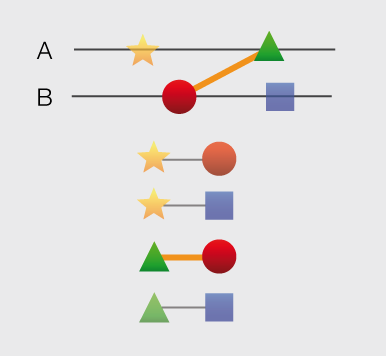
In this case we select all of them and we take one template for every domain-domain pair. The name of the domains used in the modeling of the interaction can be found in the table of structures/models for the interaction (identified by the number (3) in the image). By hovering over the lines in this pane you get a description of the PDB entry from which the structure or the model were taken. By clicking you can select the corresponding structure in the table below. |

3 |
Structures and models table This table contains details for the different structures and models for the interaction. It reports, for example, the type of structure/model, the PDB ID from which it was taken or from which the template was extracted, the starting and ending modeled residues, etc... By clicking on the View button (4) you can get a preview of the structure in the Jmol view (11) at the bottom right of the page. You can also select a set of models using the checkboxes (5) and download the corresponding PDB files (6). The different structures/models are ranked based on their quality/completeness. Please refer to this description for details on how the ranking is done. |
7 |
Interaction evidence table This table contains a list of the experimental evidences for the interaction and the references in the original databases from which the interaction was compiled. When possible a direct link to the original protein-protein interaction database and to the original publication is also provided. By clicking on the PSICQUIC link below the table (8) it is also possible to use the interaction for querying the PSICQUIC View service provided by the EBI. |
9 |
Interaction neighborhood This pane presents a quick view of the interaction neighborhood. It contains all the neighboring interactions that involve the two partners of the current interaction. You can browse and click on it just like you do on the network visualization of the Network page. By clicking on the link at the bottom of the network view (10) you can go back to the Network page and visualize the entire network from which you started your browsing (the one you obtained originally by querying Interactome3D with a list of proteins of your interest or by browsing it using GO terms). |
11 |
Jmol view The Jmol applet on the bottom right of the page shows a preview of the structure/model for the interaction. The two proteins are colored in orange and blue. In case of multiple structures/models you can change the one that is visualized by clicking on the "View" buttons (4) of the Structures and models table (3). |
Data for pre-built and user datasets calculated by Interactome3D can be downloaded for offline analysis. When you click on the download button you will be taken to a page where you can download two different sets of results:
- Complete set: This set contains all the structures and models that have been collected and generated for both proteins and interactions
- Representative set: This set contains only the top ranked structures and models that have been collected and generated for both proteins and interactions
Both in the complete and in the representative set, the same protein can have more than one structure or model. Structures for proteins and interactions are collected from the Protein Data Bank (PDB) while homology models for single proteins are collected from Modbase and homology models for interactions are generated using Modeller on structural templates extracted either from PDB biological unit files or domain-domain structural templates downloaded from 3did.
Stuctures and models for the same protein or interaction are ranked based on several criteria. You can see the ranking of the structures in the table contained in the pages showing details for proteins and interactions when browsing the results on-line. The ranking has the form n.m where n is the major rank index while m is the minor one. Structures with lower rank major index are, in general, of a higher quality then structures with a higher rank major index. The criteria used to rank structures and models for single proteins are the following:
- Complete experimental structures are ranked first in the list and in order of decreasing coverage. A structure is considered to be complete if it covers more than 80% of the protein sequence length. A complete experimental structure always has the rank minor index equal to 0.
- Complete homology models are ranked just after complete experimental structures in decreasing order of sequence identity of the template to the target. A complete homology model always has the rank minor index equal to 0.
- Partial experimental structures and
homology models are ranked after complete homology
models. In this case Interactome3D tries to group together
structures that cover, together, the largest portion of
the protein sequence length. The different groups are
generated by a greedy algorithm that takes into
consideration the structures sorted by decreasing values
of the following scoring function:
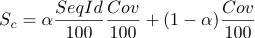
which takes into account the coverage of the structure and the sequence identity of the template to the target protein. The parameter alpha is set to 0.95 from empirical observations. Every group of structures/models is assigned the same rank major number and different rank minor numbers in order of starting residue of the structure.
When creating the representative set only the structures/models with rank major index equal to 1 are retained. This means that also in the representative set we can have more than one structure for the same protein. This happens when a protein only has structures/models with a coverage lower than 80%. In this case, as described before, the program selects a group of (potentially) overlapping structures/models that together span the largest portion of the protein sequence.
The ranking for interaction structures/models is simpler:
- Experimental structures are ranked
first in the list and in decreasing order of the values of
the following score function
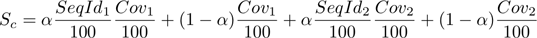
where the sequence identity and coverage of the two interacting proteins are taken into consideration. The parameter alpha is set to 0.95 from empirical observations. - Homology models from generic PDB templates are ranked after the experimental structures in decreasing order of the values of the previous score function
- Homology models from domain-domain structural templates are ranked at the end of the list in decreasing order of the values of the previous score function
In the case of interactions the representative dataset contains only one structure per interaction, the one with rank major number equal to 1. The rank minor index for interaction structures/models is always equal to 0.
For every set of results (complete and representative) there are two tables (proteins.dat and interactions.dat) summarizing the structures and models collected and generated for proteins and interactions and two sets of tarballs (proteins_xxx.tgz and interactions_xxx.tgz) containing the corresponding PDB files. Every tarball contains a maximum of 500 structures. If the dataset contains more than 500 structures they are splitted into different tarballs named with an increasing numbering scheme.
The table proteins.dat contains, for every structure, the following columns:
| # | Name | Description |
|---|---|---|
| 1 | UNIPROT_AC | Uniprot AC of the corresponding protein |
| 2 | RANK_MAJOR | Rank major index |
| 3 | RANK_MINOR | Rank minor index |
| 4 | TYPE | Type of the structure. Can be either "Structure" or "Model". |
| 5 | PDB_ID | The PDB id of the experimental structure or the template used for the modeling. |
| 6 | CHAIN | The chain id of the experimental structure or the template used for the modeling. |
| 7 | SEQ_IDENT | The sequence identity between the structure and the target protein (in percentage). Please notice that, due to possible mutations present in the crystallized protein the sequence identity for experimental structures could be lower than 100%. |
| 8 | COVERAGE | The coverage of the target protein by the structure or model (in percentage). This corresponds to the length of the structure divided by the length of the protein sequence. |
| 9 | SEQ_BEGIN | The first residue in the protein sequence aligned with the experimental structure or the first modeled residue of the target protein. Please notice that, since all the chain in the PDB file is taken (and not only the part of the chain actually aligned by BLAST) there could be differences in the actual correspondence of the starting/ending points reported by the table. |
| 10 | SEQ_END | The last residue in the protein sequence aligned with the experimental structure or the last modeled residue of the target protein. Please notice that, since all the chain in the PDB file is taken (and not only the part of the chain actually aligned by BLAST) there could be differences in the actual correspondence of the starting/ending points reported by the table. |
| 11 | GA431 | For homology models the GA431 score reported by ModPipe. |
| 12 | MPQS | For homology models the MPQS score reported by ModPipe. |
| 13 | ZDOPE | For homology models the ZDOPE score reported by ModPipe. |
| 14 | FILENAME | The name of the PDB file containing the structure/model for the protein. It refers to the files contained in the tarballs proteins_xxx.tgz. |
The table interactions.dat contains, for every structure, the following columns:
| # | Name | Description |
|---|---|---|
| 1 | PROT1 | Uniprot AC of the first interacting protein |
| 2 | PROT2 | Uniprot AC of the second interacting protein |
| 3 | RANK_MAJOR | Rank major index |
| 4 | RANK_MINOR | Rank minor index |
| 5 | TYPE | Type of the structure. Can be one of "Structure", "Model" or "Dom_dom_model". |
| 6 | PDB_ID | The PDB id of the experimental structure or the template used for the modeling. |
| 7 | BIO_UNIT | Experimental structures for interactions, as well as template from generic PDB files (excluding domain-domain structural templates from 3did) are taken from PDB biological units. This field contains the name of the PDB file of the corresponding biological unit. See the PDB website for a description of biological units files. |
| 8 | CHAIN1 | The chain id corresponding to the first interacting protein. |
| 9 | MODEL1 | The MODEL id corresponding to the first interacting protein. Biological units files, in fact, may contain more than one MODEL. See the PDB website for a description of biological units files. |
| 10 | SEQ_IDENT1 | The sequence identity between the template/structure and the first interacting protein (in percentage). Please notice that, due to possible mutations present in the crystallized protein the sequence identity for experimental structures could be lower than 100%. |
| 11 | COVERAGE1 | The coverage of the first interacting protein by the template/structure (in percentage). This corresponds to the length of the structure divided by the length of the protein sequence. |
| 12 | SEQ_BEGIN1 | The first residue in the protein sequence of the first interacting protein aligned with the experimental structure (or modeled). Please notice that, since for experimental structures all the chain in the PDB file is taken (and not only the part of the chain actually aligned by BLAST) there could be differences in the actual correspondence of the starting/ending points reported by the table. |
| 13 | SEQ_END1 | The last residue in the protein sequence of the first interacting protein aligned with the experimental structure (or modeled). Please notice that, since for experimental structures all the chain in the PDB file is taken (and not only the part of the chain actually aligned by BLAST) there could be differences in the actual correspondence of the starting/ending points reported by the table. |
| 14 | DOMAIN1 | For models obtained from domain-domain structural templates this is domain in the first interacting protein used for modeling the interaction. |
| 15 | CHAIN2 | The chain id corresponding to the second interacting protein. |
| 16 | MODEL2 | The MODEL id corresponding to the second interacting protein. Biological units files, in fact, may contain more than one MODEL. See the PDB website for a description of biological units files. |
| 17 | SEQ_IDENT2 | The sequence identity between the template/structure and the second interacting protein (in percentage). Please notice that, due to possible mutations present in the crystallized protein the sequence identity for experimental structures could be lower than 100%. |
| 18 | COVERAGE2 | The coverage of the second interacting protein by the template/structure (in percentage). This corresponds to the length of the structure divided by the length of the protein sequence. |
| 19 | SEQ_BEGIN2 | The first residue in the protein sequence of the second interacting protein aligned with the experimental structure (or modeled). Please notice that, since for experimental structures all the chain in the PDB file is taken (and not only the part of the chain actually aligned by BLAST) there could be differences in the actual correspondence of the starting/ending points reported by the table. |
| 20 | SEQ_END2 | The last residue in the protein sequence of the second interacting protein aligned with the experimental structure (or modeled). Please notice that, since for experimental structures all the chain in the PDB file is taken (and not only the part of the chain actually aligned by BLAST) there could be differences in the actual correspondence of the starting/ending points reported by the table. |
| 21 | DOMAIN2 | For models obtained from domain-domain structural templates this is domain in the second interacting protein used for modeling the interaction. |
| 22 | FILENAME | The name of the PDB file containing the structure/model for the interaction. It refers to the files contained in the tarballs interactions_xxx.tgz. |
Feel free to contact us if you have any question about the format of the downloaded data and how to use them.
The data contained in Interactome3D can be accessed programmatically through a RESTful interface. The interface is available through the 'api' subfolder and the URL of the request is encoded as in the following examples:
https://interactome3d.irbbarcelona.org/api/getProteinStructures?uniprot_ac=A0A5B9 https://interactome3d.irbbarcelona.org/api/getInteractionStructures?queryProt1=A0A5B9&queryProt2=P01848
The following functions are available:
- getUniprotACs It returns the Uniprot ACs of all the proteins contained in one of the precalculated datasets in Interactome3D. It takes no parameters and returns the list of Uniprot ACs as raw text, separated by newlines.
- getProteinStructures It returns the list of structures for
one protein . It takes only
one parameter:
- uniprot_ac The Uniprot AC of the protein of interest
- getInteractionStructures It returns the list of structures for
one interaction. It takes two parameters:
- queryProt1 The Uniprot AC of the first protein
- queryProt2 The Uniprot AC of the second protein
- getPdbFile It returns the contents of one of the PDB files
contained in Interactome3D. It takes two parameters:
- filename The name of the PDB file as contained in the proteins.dat or interactions.dat file or returned by one of the previous APIs
- type Can be either protein or interaction depending if you want to fetch the PDB file of a single protein or interaction.
- getVersion It returns the current version of Interactome3D, as raw text.
- How frequently is Interactome3D updated?
- What is the difference between complete structures, complete models and partial models for proteins?
- If I query Interactome3D with proteins X, Y and Z the answer includes proteins X, T, W but no Y and no Z. Why is the set of query proteins so different from the answer?
- If I query Interactome3D with proteins A, B and C the answer includes many more proteins. How can I restrict the results to only the proteins in specified in the query?
- What is the meaning of the blue bar (available for docking) in the interactions chart in the results page?
How frequently is Interactome3D updated?
Twice a year, every 6 months.
What is the difference between complete structures, complete models and partial models for proteins?
Structures and models are considered to be complete if they span at least 80% of the length of the protein sequence. Otherwise they are classified as partial.
If I query Interactome3D with proteins X, Y and Z the answer includes proteins X, T, W but no Y and no Z. Why is the set of query proteins so different from the answer?
The pre-built datasets contained in Interactome3D are composed of binary interactions available in public Databases (Intact, MINT, HPRD, BIND, etc...) and the corresponding structural data available in the PDB or that can be modeled by homology. When the user queries Interactome3D with a set proteins the result of the query will be all the interactions involving at least one of the proteins in the query set. In the case of the example Interactome3D contains experimentally identified binary interactions only for protein X (interacting with proteins T and W). No interactions have been discovered (or at least are not available in the public databases that Interactome3D uses) for the other two query proteins (Y and Z). For this reason the answer will include the proteins that are known to interact with X (T and W) but not the query proteins Y and Z.
If I query Interactome3D with proteins A, B and C the answer includes many more proteins. How can I restrict the results to only the proteins in specified in the query?
By default, Interactome3D returns all the interactions involving any protein in the query list. If you want to only display interactions between any two proteins in your query list you can check the option "Only show the proteins in the list" below the input box in the home page. If you do so, the results will include only the interactions for which both the interactors are in your query list. This is particularly useful if you want to visualize the experimentally validated interactions between components of the same complex.
What is the meaning of the blue bar (available for docking) in the interactions chart in the results page?
For those interactions that do not have a structure
or a model there is still hope, provided that we have a structure or a
model for both the single interactors. These are exactly the
interactions that correspond to the blue portion of the bar. In these
cases, we can apply computational protein-protein docking in order to
try to predict the structure of the interaction.
The current version of Interactome3D does not automatically apply
docking to those cases but we have an internal pipeline that can be
used for running docking experiments in a high-throughput manner on
all the interactions having structures/models for both the unbound
proteins.
Due to the computational complexity of the problem we will take into
consideration the possibility of applying such protocol on a
collaboration basis.
Interactome3D is provided by the Structural
Bioinformatics and Network Biology Group at the Institute for
Research in Biomedicine. Please contact
interactome3d![]() irbbarcelona.org for any question/suggestion.
Last modified: 28 September 2017.
irbbarcelona.org for any question/suggestion.
Last modified: 28 September 2017.